Product Features
A better way to explore quantum computing
Turn-key Implementations
For classically trained data scientists that want to test what is possible and experts that are benchmarking the state-of-the-art perfomance – this is the place to start.
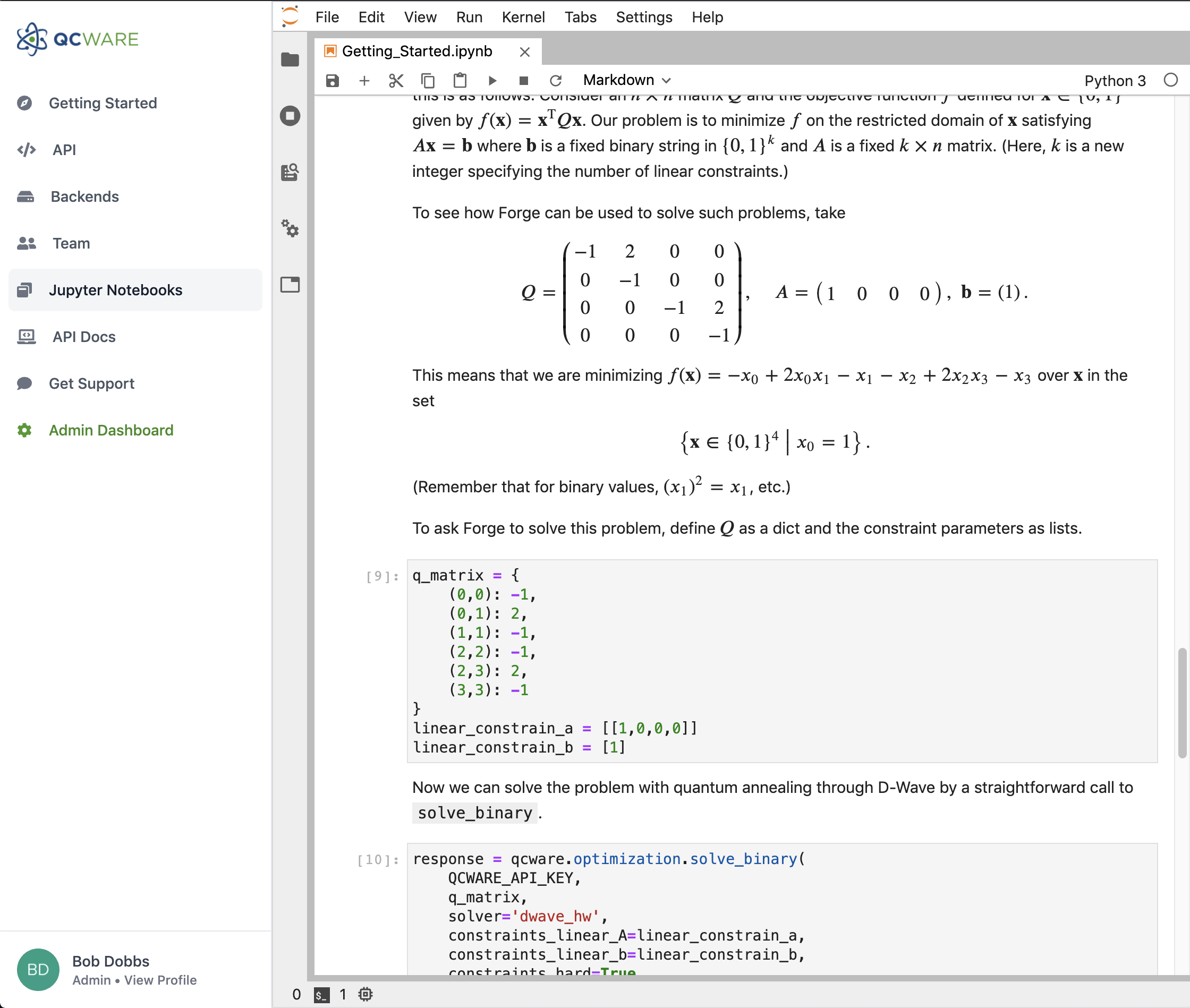
Binary Optimization
Get a significant performance boost for D-Wave hardware and use the latest improvements for gate-based approaches.
Machine Learning & A.I.
Try out quantum data loaders and algorithms with guaranteed speed-ups on clustering, classification, and regression.
Chemistry Simulation
Find ground and excited states for photoactive molecules. Calculate gradients and couplings of large molecules.
Blazing fast development and testing
Import your existing circuits, edit, and simulate them. Use GPUs on the cloud to get results of large quantum circuits in a matter of minutes.
GPU-accelerated simulation
Your simulation runs on GPUs provisioned on the cloud. A 20 qubit circuit with 1000 gates takes about 6 seconds of GPU-time to execute.
- Ψ
Custom Circuits
Design, test, and run custom quantum circuits on gate-model simulators or hardware from companies like Rigetti and IonQ with quasar.
Only pay for what you use
Pay-as-you-go, and pricing packages appropriate for small labs and large enterprises alike.
Ready to dive in?
Start your free trial today.
⭐ Private Beta ⭐
Chemistry Simulation API
Are you struggling with classical DFT calculations? Use the most advanced quantum computing API that works with large molecules in your quantum chemistry workflow.
Photoactive Molecules
Use a combination of classical preprocessing techniques (AEIM) and the latest in quantum algorithms (MC-VQE) to calculate ground and excited states for large photoactive molecules.
Observable Properties
Compute arbitrary observable properties, including response properties. This tool allows for the prediction of optical and electronic characteristics of chemical systems.
Gradients and Couplings
Compute force vectors and non-adiabatic coupling vectors on the nuclei of large molecular systems. Crucial for the ability to do geometry optimizations, transition paths, and dynamics simulations on interesting chemical phenomena.
Flexible licensing for your environment
We work where you work — deploy our solution in your datacenter or in the cloud service of your choice. We offer flexible licensing plans to fit any workflow.

We’re here to help
Not sure if you're ready to jump in? Need to augment your research team with a partner? Need help with a custom use case or algorithm? Contact us any time to speak with a member of our team who can help.